
Why AUGMENT® Regenerative Solutions?
The Fusion Solution in Hindfoot and Ankle
AUGMENT® Regenerative Solutions is a ground breaking technology in foot and ankle in arthrodesis surgery. The product is the first and only FDA approved alternative to autograft in ankle and hindfoot arthrodesis. Literature reports a range of 10% to as high as 41% of all hindfoot and ankle fusions fail to launch.* Can you predict which of your patients will result in a non-union?
*Haddad SL, Coetzee JC, Estok R, Fahrbach K, Banel D, Nalysnyk L. Intermediate and long-term outcomes of total ankle arthroplasty and ankle arthrodesis. A systematic review of the literature. J Bone Joint Surg Am. 2007 Sep;89(9):1899-905.
A Better Way.
-
Proven
- Level 1 evidence from largest F&A clinical trial ever conducted
- Equivalent improvements in clinical outcomes, compared to the gold standard autograft1
-
Unique
- Bioengineered human PDGF-BB stimulates multiple aspects of healing in response to injury
- rhPDGF-BB is highly purified with consistent biological potency; allograft and autograft are highly variable in quality and potency2,3,4,5
-
Labeled
- Class III combination product labeled for ankle and hindfoot arthrodesis
-
Safe
- In commercial use since 2009 (Canada)
- Eliminates risks, morbidities, costs associated with autograft harvest
2. Fiedler, et al. J Cell Biochem (2002).
3. Ozaki, et al. J Stem Cells & Dev (2007).
4. Bouletreau, et al. Plast Reconstr Surg (2002).
5. Hollinger, et al. JBJS (2008).
![]()
Both AUGMENT® Bone Graft and AUGMENT® Injectable are made up of two components.
- rhPDGF-BB, a man-made protein called recombinant human platelet-derived growth factor (rhPDGF-BB). It is manufactured from yeast cells and has been approved by the FDA for more than 20 years.
- The other part is a bone-like substance called beta-Tricalcium Phosphate (β-TCP), which has been approved by the FDA for more than 32 years for AUGMENT® Bone Graft and includes a collagen matrix to make AUGMENT® Injectable flowable.


Preparing AUGMENT® Bone Graft
AUGMENT® Bone Graft is supplied as a two component kit. Each kit consists of:
- A cup containing β-TCP particulate
- A vial containing solution of 0.3 mg/ml rhPDGF-BB in sodium acetate buffer
NOTE: Preparation of this mixture should be done at least 10 minutes before implantation in the joint space(s) intended for arthrodesis because it requires this time period to saturate. Please plan accordingly.

Using sterile technique, transfer both the cup (containing the β-TCP granules) as well as the vial (containing the rhPDGF-BB solution) to the sterile field.

First open the cup and transfer the β‑TCP granules to a separately available sterile surgical bowl.

Then using a syringe and needle, draw up the liquid contents of the vial in its entirety (the rhPDGF-BB solution). Transfer all of the fluid to the surgical bowl containing the β-TCP granules.
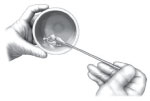
Using a spatula, curette, or similar instrument, gently stir these two components together for approximately 30 seconds to ensure a homogeneous mixture. This mixture should, at that point, have the consistency of wet sand.

The rhPDGF-BB saturated graft mixture should be left undisturbed for 10 minutes before being implanted to ensure optimal saturation of the β-TCP particles. Ensure that the entire volume of both components is combined. The product should be implanted within one (1) hour of mixing the two components.

Immediately prior to implantation, the entire contents should be mixed briefly again to ensure complete saturation of the β-TCP particles.
AUGMENT® Bone Graft should be implanted on already prepared host bone surfaces, being careful not to overstuff the joint space(s). This material should be inserted amongst all peri-articular defects (both pre-existent and surgically created). This will maximize bony apposition but not impede direct host bone to host bone apposition.
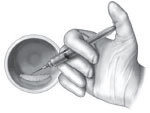
Any rhPDGF-BB liquid remaining in the bowl after implantation of a sufficient amount of AUGMENT® Bone Graft may then be drawn up and used to hydrate the already implanted AUGMENT® Bone Graft dispersed throughout the fusion site.
Applying AUGMENT® Bone Graft
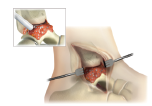
Irrigate and then remove all fluid from the surgical site one final time after joint preparation is complete and immediately prior to AUGMENT® Bone Graft implantation.
Assess the fusion site. Determine where all the bony defects (e.g., subchondral voids and surface irregularities) are which will need to be filled with AUGMENT® Bone Graft.
Manually pack AUGMENT® Bone Graft into, not around, all these bony defects throughout the joint(s) intended for arthrodesis. Be sure to place AUGMENT® Bone Graft wherever there is not direct host bone to host bone apposition.
Care should be taken to ensure that all AUGMENT® Bone Graft material is contained within the perimeter of these bony defects and that the graft remains saturated during the surgical procedure.
REMEMBER: It is important to ensure that all bony defects are grafted. Adequate graft fill is needed to optimize results with any grafting material.
NOTE: Overfilling or “overstuffing” of the joint space with this material is unnecessary, and should be avoided, particularly to the extent that direct host bone to host bone apposition is prevented. In order to achieve adequate closure and containment of the material, however, it is imperative that the material be contained with a meticulous soft tissue and capsular closure. This will keep the material from the surrounding soft tissue, in order to achieve appropriate containment within the desired peri-articular site.
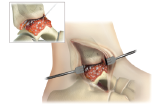
Hydrate the implant site with any remaining rhPDGF-BB solution, if desired.
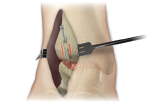
Reduce the joint and apply rigid fixation.
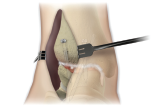
Following satisfactory joint reduction and hardware fixation of the arthrodesis site(s), any remaining (unused) AUGMENT® Bone Graft should be packed around the external perimeter of the treated joint(s).
Preparing AUGMENT® Injectable
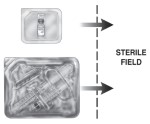
Using sterile technique, transfer the vial and matrix tray components to the sterile field.
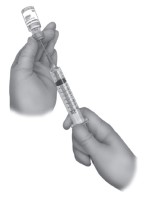
Completely withdraw the contents of the vial containing the rhPDGF-BB solution using the empty syringe and 18G beveled needle. Displace any air remaining in the syringe.
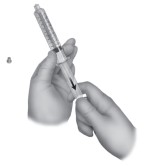
Remove the cap from the syringe containing the β-TCP/ collagen matrix. Pull the plunger to the 10mL mark and tap the syringe barrel to loosen the matrix.

Connect the syringe containing the rhPDGF-BB solution with the syringe containing the matrix using the female-to-female luer lock connector.

Transfer the rhPDGF-BB solution into the syringe containing the matrix. After transferring all the rhPDGF-BB solution, pull the plunger on the syringe containing the hydrated matrix past the 10mL mark and release. Let the syringes sit undisturbed for a minimum of 90 seconds.
Note: For the 1.5cc kit, pull the plunger to the 5.5mL mark.

After hydrating the matrix, transfer the contents back and forth between the two syringes for no less than twenty (20) cycles to form a homogeneous paste. Note: A cycle is defined as passing the matrix to the empty syringe and back.

Transfer all the paste to one of the syringes, then crack (but do not fully release) the connection between the syringe containing the paste and the female-to-female luer lock connector, while simultaneously gently pulling the plunger on the syringe containing the paste, to relieve any pressure built up during the mixing process.

Disconnect the empty syringe and female-to-female luer lock connector from the syringe containing the paste.
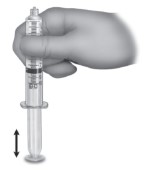
Hold the syringe barrel and gently tap (vertically) onto the plunger end to shift the paste toward the plunger seal, then express any excess air (without displacing product).

Connect the 14G blunt cannula and dispense the paste into the graft site.
Applying AUGMENT® Injectable
Using the prepared syringe (see AUGMENT® Injectable Preparation section, above), dispense the hydrated matrix into the void. Note: It may require an initial force to get the paste to flow through the 14 gauge needle. However, once the paste starts to flow, the force required to maintain a flow will be reduced.
Carefully apply the hydrated matrix to the surgical site (i.e. the subchondral voids, and surface irregularities visualized throughout the entire joint) such that the graft material is in contact with the entire osseous surfaces to be fused/repaired while allowing rigid fixation and primary bone contact to occur between the osseous surfaces. Immediately after joint reduction and hardware fixation of the fusion site, any remaining (unused) AUGMENT® Injectable should be applied around the external perimeter of the fusion construct.
- Because AUGMENT® Injectable does not harden or “set”, standard hardware implantation to ensure rigid fixation is necessary.
- In order to enhance the formation of new bone, AUGMENT® Injectable
should be placed in direct contact with well-vascularized bone. Cortical bone may be perforated prior to placement of the AUGMENT® Injectable material.
Remember: It is important to ensure that all bony defects are grafted. Adequate graft fill is needed to optimize results with any grafting material.
Once AUGMENT® Injectable has been applied to the defect site, carefully layer periosteal and overlying soft tissue to enclose and contain the graft material.
Care should be taken not to irrigate the graft site following implantation of AUGMENT® Injectable.
- To guard against ectopic bone formation, take care to prevent AUGMENT® Injectable extrusion beyond the desired fusion regions, especially during hardware placement and joint reduction.
Any remaining graft material should be discarded.
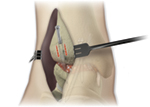
Reduce the joint and apply rigid fixation.
Indications for Use
AUGMENT® Bone Graft and AUGMENT® Injectable are indicated for use as an alternative to autograft in arthrodesis (i.e., surgical fusion procedures) of the ankle (tibiotalar joint) and/or hindfoot (including subtalar, talonavicular, and calcaneocuboid joints, alone or in combination), due to osteoarthritis, post-traumatic arthritis, rheumatoid arthritis, psoriatic arthritis, avascular necrosis, joint instability, joint deformity, congenital defect, or joint arthropathy in patients with preoperative or intraoperative evidence indicating the need for supplemental graft material.
CONTRAINDICATIONS
AUGMENT® Bone Graft and AUGMENT® Injectable should not:
- be used in patients who have a known hypersensitivity to any of the components of the product or are allergic to bovine collagen (AUGMENT® Injectable only) or yeast-derived products.
- be used in patients with active cancer.
- be used in patients who are skeletally immature (<18 years of age or no radiographic evidence of closure of epiphyses).
- be used in pregnant women. The potential effects of rhPDGF-BB on the human fetus have not been evaluated.
- be implanted in patients with an active infection at the operative site.
- be used in situations where soft tissue coverage is not achievable.
- be used in patients with metabolic disorders known to adversely affect the skeleton (e.g. renal osteodystrophy or hypercalcemia), other than primary osteoporosis or diabetes.
- be used as a substitute for structural graft.
Warnings
As with all therapeutic recombinant proteins, there is a potential for immune responses to be generated to the rhPDGFBB component of AUGMENT® Bone Graft and AUGMENT® Injectable. The immune response to rhPDGF-BB was evaluated for AUGMENT® Injectable in two studies, and for AUGMENT® Bone Graft (which contains the identical rhPDGF-BB) in two pilot and one pivotal study for ankle and hindfoot arthrodesis procedures. The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies to AUGMENT® Bone Graft or AUGMENT® Injectable with the incidence of antibodies to other products may be misleading.
Women of childbearing potential should avoid becoming pregnant for one year following treatment with AUGMENT® Bone Graft or AUGMENT® Injectable. The implantation of rhPDGF-BB in women and the influence of their development of anti-PDGF-BB antibodies, with or without neutralizing activity, on human fetal development are not known.
The safety and effectiveness of AUGMENT® Bone Graft or AUGMENT® Injectable in nursing mothers has not been established. It is not known if rhPDGF-BB is excreted in human milk.
The safety and effectiveness of AUGMENT® Bone Graft or AUGMENT® Injectable has not been established in anatomical locations other than the ankle or hindfoot, or when combined with autologous bone or other bone grafting materials.
The safety and effectiveness of repeat applications of AUGMENT® Bone Graft or AUGMENT® Injectable have not been established.
The safety and effectiveness of AUGMENT® Bone Graft or AUGMENT® Injectable in pediatric patients below the age of 18 years have not been established.
AUGMENT® Bone Graft or AUGMENT® Injectable do not have any biomechanical strength and must be used in conjunction with standard orthopedic hardware to achieve rigid fixation.
The β-TCP component is radiopaque, which must be considered when evaluating radiographs for the assessment of bridging bone. The radiopacity may also mask underlying pathological conditions. Over time, the β-TCP is intended to be resorbed at the fusion site and replaced by new bone. Under such circumstances, it would typically be indistinguishable from surrounding bone.
Please refer to the full package insert for more information:
AUGMENT® Bone Graft
AUGMENT® Injectable
SUPPORTING CLINICAL EVIDENCE
- DiGiovanni, CW et al. | J Bone Joint Surg Am. 2013

- Friedlaender, GE et al. | Curr Pharm Des. 2013
- Solchaga, LA et al. | Clin Drug Investig. 2013
- Glazebrook, M et al. | Techniques in Foot & Ankle Surgery 2012
- DiGiovanni, CW et al. | Expert Rev Med Devices 2012
- DiGiovanni, CW et al. | Foot Ankle Int. 2011
- DiGiovanni, CW et al. | Foot Ankle Clin. 2010
- Daniels, T et al. | Foot Ankle Int. 2010
- Hollinger, JO et al. | J Bone Joint Surg Am. 2008
- Nevins, M et al. | J Periodontol 2005
- Solchaga, LA et al. | J Tissue Eng. 2012
- Caplan, AI et al. | J Orthop Res. 2011
- Verma, R et al. | Current Ortho Practice 2011
- Pountos, I et al. | J Orthop Trauma 2010
- Moore, DC et al. | J Bone Joint Surg Am. 2009
- Al-Zube, L et al. | J Orthop Res. 2009
- McCarthy, HS et al. | J Cell Physiol. 2009
- Tokunaga, A et al. | J Bone Miner Res. 2008
- Sanchez-Fernandez, MA et al. | PLoS One. 2008
- Hollinger, JO et al. | J Orthop Res. 2008
- Ozaki, Y et al. | Stem Cells Dev. 2007
- Kilian, O et al. | Growth Factors 2005
- Fiedler, J et al. | J Cell Biochem. 2004
- Mehrotra, M et al. | J Cell Biochem. 2004
- Kilian, O et al. | Eur J Med Res. 2004
- Bouletreau, PJ et al. | Plast Reconstr Surg. 2002
- Fiedler, J PJ et al. | J Cell Biochem. 2002
- Zhang, Z PJ et al. | Biochem Biophys Res Commun. 1998
- Glazebrook, M PJ et al. | Foot Ankle Int. 2013
- Baumhauer, JF PJ et al. | Foot Ankle Int. 2013
- Baumhauer, J PJ et al. | Foot Ankle Int. 2013
- DiGiovanni, CW et al. | J Bone Joint Surg Am. 2016
- LIN, SS ET AL. | J
AM ACAD ORTHOP SURG. 2016
- KRAUSE, F ET AL. |
JBJS 2016
- DiGiovanni, CW et al. | J Bone Joint Surg Am. 2013